
What's the difference between proving a medical device's equivalence in the EU and the US?
Find out how these distinct standards impact our industry. Let's dive in! 👇


The world of medical device regulation is complex, with significant differences between the EU's and the US's approaches to proving equivalence.
1) EU's MDR: Clinical, Technical, and Biological Equivalence :
In the EU, the Medical Device Regulation (MDR) outlines stringent requirements for proving equivalence, as detailed in the MDCG 2020-5 Guidance on Clinical Evaluation - Equivalence.
This document lays out specific criteria across three key domains: technical, biological, and clinical characteristics.
Technical Characteristics:
According to the MDR, the device must be of similar design, used under similar conditions, have similar specifications and properties (like intensity of energy, tensile strength, viscosity, surface characteristics, wavelength, and software algorithms), employ similar deployment methods, and have similar principles of operation and critical performance requirements.
Example: For a new cardiac pacemaker, the EU demands that it matches an existing product in aspects like energy output, the material of leads, and software algorithms governing heart rate modulation.
Biological Characteristics:
The MDR stipulates that the device uses the same materials or substances in contact with the same human tissues or body fluids, for a similar kind and duration of contact, and exhibits similar release characteristics of substances, including degradation products and leachables.
Example: A new orthopedic implant would need to use the same alloy as an existing implant, ensuring similar biocompatibility and degradation rates.
Clinical Characteristics:
The device should be used for the same clinical condition or purpose, with similar severity and stage of disease, at the same site in the body, in a similar population (considering age, anatomy, physiology), and have the same kind of user. It also should demonstrate similar relevant critical performance in view of the expected clinical effect for a specific intended purpose.
Example: A new type of insulin pump must demonstrate efficacy in controlling blood glucose levels in similar patient demographics as an existing pump, ensuring it is suitable for the same age group and disease severity.
For a structured pathway to equivalence, consult the accompanying table from the MDCG guidance:
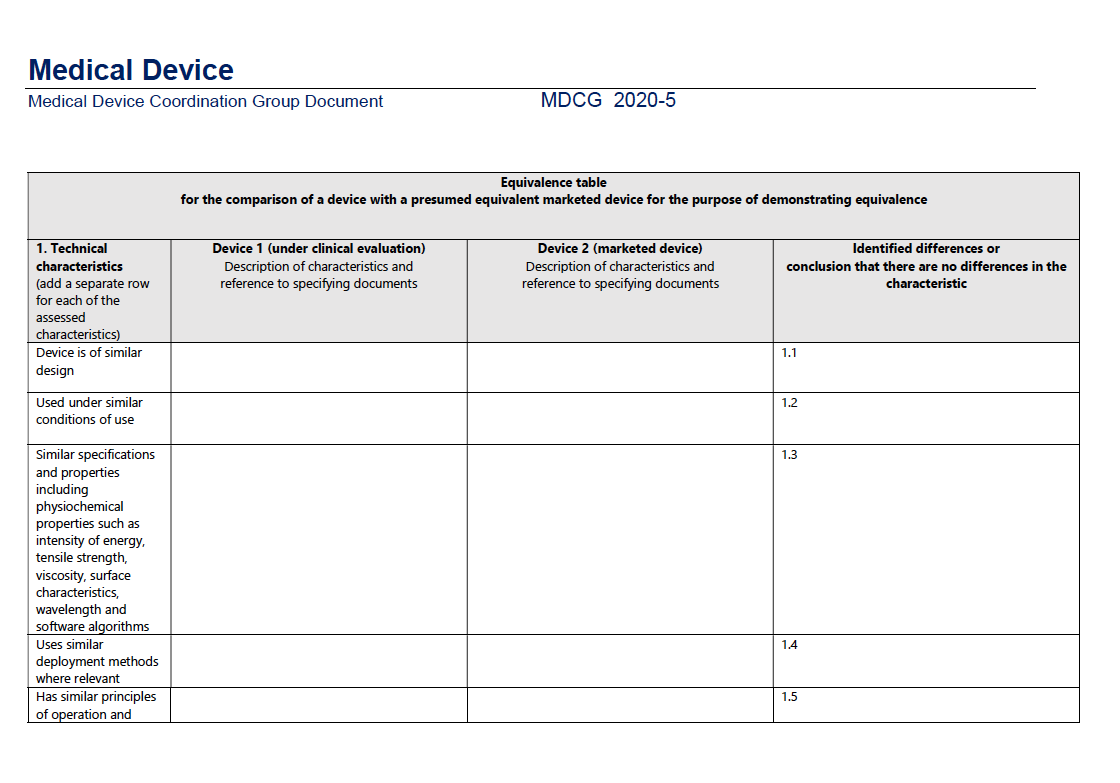
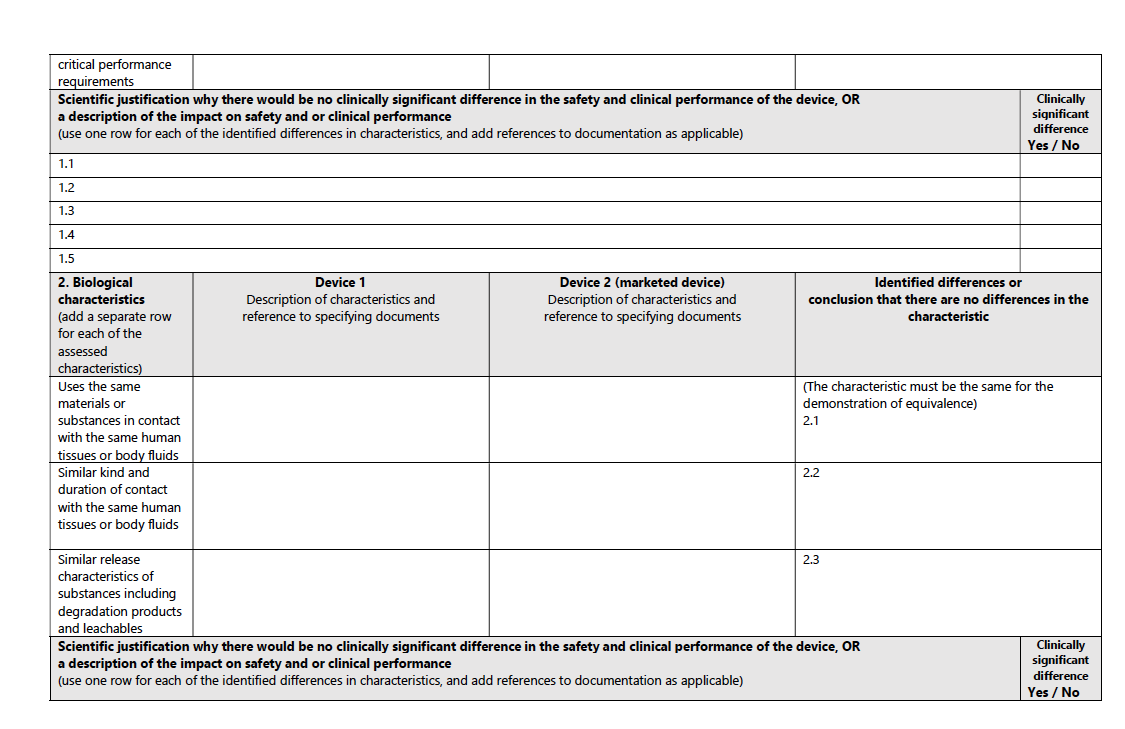
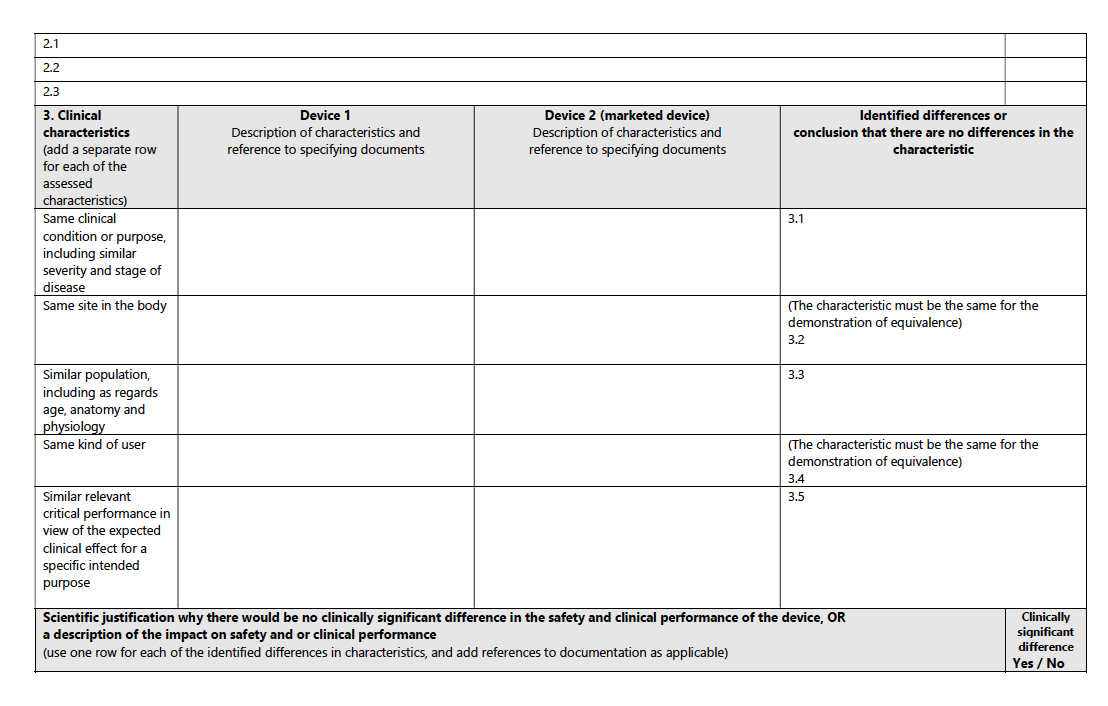

2) US FDA: Substantial Equivalence:
In contrast to the EU's approach, the US FDA's framework for determining substantial equivalence offers a different paradigm, which can be better understood through their guidance on evaluating substantial equivalence in premarket notifications 510(k).
The FDA's standard for substantial equivalence hinges on two key aspects:
Same Intended Use as the Predicate:
The new device must have the same intended use as the predicate device. This means it should be used for the same medical purpose and conditions as the already marketed device.
Technological Characteristics:
- Same Technological Characteristics:
If the new device has identical technological characteristics as the predicate, it could be deemed substantially equivalent.
- Different Technological Characteristics but As Safe and Effective:
If the new device has different technological characteristics, it can still qualify for substantial equivalence if it is demonstrated to be as safe and effective as the predicate. Importantly, it should not raise different questions of safety and effectiveness compared to the predicate device.
Example:
Imagine a company developing a laser surgery tool for eye treatments. The FDA would evaluate whether this new device has the same purpose (e.g., correcting vision) as an existing laser tool. If the new tool uses a different type of laser technology, the company must provide evidence showing that this new technology is as safe and effective as the existing one, without introducing new safety concerns.
Balancing Safety with Technological Advancement:
The FDA's approach, as delineated in their guidance, balances the need for maintaining safety and effectiveness with the opportunity for technological innovation. This framework allows for introducing new technologies in medical devices, provided they meet stringent safety standards and demonstrate effectiveness comparable to existing devices.
3) Comparative Analysis: Is It Justifiable?
Is comparing these two approaches directly beneficial or even appropriate?
The EU's stringent, multi-faceted criteria reflect a comprehensive approach to equivalence, emphasizing detailed conformity in multiple aspects. In contrast, the US FDA's standard allows for technological innovation, provided the core aspects of safety and efficacy are maintained.
This contrast in regulatory outlook is pivotal for medical device professionals formulating market entry strategies. Though patient safety and device efficacy are shared goals, the methods to establish them differ markedly between the EU and the US. The FDA's equivalence procedure and the EU's approach serve distinct objectives within their regulatory frameworks: the former centers on conformity assessment, while the latter emphasizes the strategic use of clinical evidence. This fundamental difference shapes the specific requirements and nuances of each process.
4 ) Conclusion:
In summary, for industry professionals, discerning the particularities of each regulatory environment is crucial. It is imperative to align product development and clinical evaluation strategies with the specific demands of each jurisdiction. By doing so, we ensure adherence to patient safety and device efficacy standards. The expertise in these regulatory distinctions will continue to be a cornerstone of successful market penetration and maintaining the integrity of healthcare innovation.
Ps: Thank you for reading our newsletter; if you found it useful, please share it with others.